Нові можливості застосування блокатора ішемічного каскаду в терапії гострого інфаркту міокарда

Резюме. Окисний стрес відіграє особливу роль при гострому коронарному синдромі (ГКС), зокрема, є провідною ланкою патогенезу реперфузійного пошкодження міокарда.
Продукти вільнорадикального окиснення можуть запускати процеси загибелі кардіоміоцитів та бути відповідальними за 50 % кінцевого розміру зони некрозу при ГКС, виникнення реперфузійних аритмій, систолічної мікросудинної дисфункції. Тому адекватна антиоксидантна терапія, спрямована на елімінацію активних форм кисню, активацію антиоксидантів, модуляцію процесів безпосередньої та відтермінованої загибелі клітин, має бути важливою складовою менеджменту ГКС.
Едаравон є найперспективнішим антиоксидантом і блокатором ішемічного каскаду завдяки своїм фармакокінетичним та фармакодинамічним особливостям.
За даними низки експериментальних та клінічних досліджень, застосування едаравону у пацієнтів з ГКС веде до зниження рівня вільних радикалів на тлі активації таких ферментів-антиоксидантів, як супероксиддисмутаза, глутатіонпероксидаза, каталаза, до збереження нормальної архітектури мітохондрій та інших клітинних структур, пригнічення експресії різноманітних протеїнів, залучених в активацію апоптозу, фероптозу та прозапальної відповіді.
На клінічному рівні едаравон сприяє зменшенню зони некрозу з вірогідним зниженням плазмового рівня КФК-МВ, запобігає розвитку аритмій та систолічної дисфункції на тлі ішемії/реперфузії.
За результатами рандомізованих плацебо-контрольованих досліджень, застосування едаравону у пацієнтів з ГКС до проведення реперфузії у перші 6 годин після виникнення симптомів є ефективним у зменшенні зони інфаркту та профілактиці реперфузійних аритмій. Таким чином, едаравон є потужним антиоксидантом і блокатором ішемічного каскаду з додатковими антиапоптотичним та антифероптотичним, антинекротичним, протизапальним ефектами, стабілізує кардіоміоцити та ендотеліальні клітини, а його призначення при ГКС є перспективною стратегією профілактики реперфузійного пошкодження міокарда зі зменшенням зони некрозу, профілактикою фатальних порушень ритму, патологічного ремоделювання міокарда та судин.
Завдяки своїм багатогранним ефектам едаравон може бути корисним і при інших кардіоваскулярних захворюваннях.
Ключові слова: гострий коронарний синдром; едаравон; антиоксидант; огляд
Дослідження Глобального тягаря хвороб (Global Burden of Disease), результати якого оприлюднені у 2020 році, вкотре підтвердило, що серцево-судинні захворювання є основними причинами смертності у країнах з різним рівнем доходу [22]. Примітно, що пандемія COVID-19 не порушила цей уже звичний порядок речей, а, навпаки, загострила проблему кардіоваскулярної захворюваності та смертності. За прогнозами фахівців ВООЗ, у 2030 році серцево-судинні захворювання заберуть 23,3 мільйона людських життів [7]. Україна не виняток, а, швидше, один із лідерів у цьому сумному списку. Як серед чоловіків, так і серед жінок основною причиною втрати років здорового життя серед кардіоваскулярних захворювань є ішемічна хвороба серця (ІХС) [7]. Хронічні та гострі форми ІХС не тільки забезпечують для нозології перші рядки у рейтингах смертності, але і збільшують частку інвалідності у глобальному тягарі хвороб, що особливо гостро відчули системи охорони здоров’я у часи пандемії.
Окисний стрес у патогенезі кардіоваскулярної патології
Ішемічна хвороба серця тісно пов’язана з такою коморбідною патологією, як атеросклероз, артеріальна гіпертензія (АГ), цукровий діабет, у першу чергу за рахунок спільних механізмів пошкодження судин та міокарда через процеси надмірного вільнорадикального окиснення. Відомо, що у патогенезі вказаних захворювань утворення надлишку активних форм кисню (АФК) відіграє важливу роль [1]. До АФК належать нестабільні молекули, які легко реагують зі складовими клітин: високоактивні радикали – гідроксильний радикал OH–, ліпідний радикал ROO–, пероксинітрит ONOO–; молекули-окиснювачі – перекис водню H2O2, синглетний кисень О2 та хлорноватиста кислота HOCl. Утворені молекули за певних умов починають реагувати між собою, що за механізмом послідовних ланцюгових реакцій веде до катастрофічного їх накопичення, яке не здатні подолати антиокиснювальні системи організму [1]. Для розуміння місця окисного стресу у патогенезі серцево-судинних захворювань необхідно детальніше розібратися з механізмами впливу його агентів на різних рівнях. Активні форми кисню утворюються у фізіологічних умовах, виконуючи роль сигнальних молекул, і лише за наявності надлишку пошкоджують компоненти клітин. На клітинному рівні АФК генеруються численними джерелами, включаючи дихальний ланцюг мітохондрій, НАДФН-оксидази, ксантиноксидази, ліпоксигенази, синтази оксиду азоту (еNOS) та циклооксигенази [4]. Існує певна ієрархія оксигенних реакцій: НАДФН-оксидази працюють на рівні плазматичної мембрани та утворюють супероксид і перекис водню, мітохондріальний ланцюг транспорту електронів генерує супероксид і перекис водню з накопиченням їх в органелі, лізосоми вивільняють реактивне залізо, яке генерує ліпідні АФК, у ядрі клітин утворюється супероксид за допомогою циклооксигеназ [19]. Запускати цей процес може ішемія, гуморальні чинники кардіоваскулярної патології, зокрема, доведено, що ангіотензин ІІ підвищує експресію НАДФН-оксидази та ксантиноксидази. Негативний вплив надлишку активних окиснювачів полягає у руйнуванні білків, ДНК, активації перекисного окиснення ліпідів у зовнішніх та внутрішніх мембранах клітин, що веде до безпосередньої або відтермінованої загибелі клітин [29]. Патологічні ефекти АФК реалізуються на клітинах різного типу, страждають ендотелій та міоцити судин, кардіоміоцити, залучаються нейтрофіли та фібробласти, для яких молекули-окиснювачі виступають хемоатрактантами. Активні форми кисню у вигляді ліпідних радикалів ROO– руйнують мембрани, порушують функціонування сигнальних білків, можуть активувати запальний каскад внутрішньоядерних сигнальних шляхів, що підтверджується зростанням рівня прозапальних цитокінів на тлі окисного стресу – фактора некрозу пухлини α (TNF-α), тромбоцитарного фактора росту (PDGF). Активація запалення – ще один наріжний камінь негативних наслідків окисного стресу, який реалізується через каскад TNF-α – нуклеарний фактор запалення В (NFκВ), з подальшою гіперпродукцією таких цитокінів, як інтерлейкіни [18]. Згадані події на клітинному та молекулярному рівні провокують безпосередню загибель клітин, запускають каскад активації сигнальних кіназ та посредників апоптозу, фероптозу, потенціюють проліферацію фібробластів та синтез колагену, що обумовлює ендотеліальну дисфункцію, патологічне ремоделювання судин та міокарда з прогресуванням кардіоваскулярної патології, розвитком фатальних ускладнень. У низці експериментальних досліджень, результати яких знаходять своє підтвердження й у клініці, показано, що надлишкове накопичення АФК може провокуватися ангіотензином ІІ через активацію НАДФ-оксидази, яка у великій кількості міститься у кардіоміоцитах, ендотеліальних та імунних клітинах. Це веде до інактивації NO-синтази з порушенням утворення NO та формуванням ендотеліальної дисфункції, до накопичення окиснених ліпідів з прогресуванням атеросклеротичного ураження, до структурного ремоделювання судин, гіпертрофії медії, до активації факторів запалення з гіперпродукцією молекул адгезії, хемокінів та активацією запальних клітин [4, 9, 17]. Розвиток ендотеліальної дисфункції, потенційований у тому числі окиснювачами, посідає чільне місце у прогресуванні основних серцево-судинних захворювань – ІХС, АГ, серцевої недостатності (СН). Показано, що індукція гіпертрофії міокарда ангіотензином ІІ реалізується також і через НАДФ-оксидазний та ксантиноксидазний механізми утворення надлишків АФК [19, 37]. За таким сценарієм відбувається ремоделювання лівого шлуночка у хворих з артеріальною гіпертензією, після інфаркту міокарда, вираженість окисного стресу визначає інтенсивність прогресування захворювання.
Загибель кардіоміоцитів під дією надлишку окиснювачів є прямим результатом окисного стресу та реалізується різними шляхами. Через пошкодження мітохондрій та інших органел відбувається безпосередня загибель клітин через некроз; шляхом активації сигнальних молекул програмується відтермінована смерть за механізмами апоптозу чи фероптозу. Причому усі варіанти загибелі клітин можуть запускатися одночасно, а АФК керують цим процесом через різні сигнальні шляхи або молекули-ініціатори. Так, апоптоз активується у зв’язку з накопиченням перекису водню та супероксиду, що вивільняє з мітохондрій фактор апоптозу AIF, який транслокується в ядро, викликає конденсацію ДНК та запускає процес загибелі [40]. Фероптоз є залізозалежним варіантом програмованої смерті клітин, пов’язаним з лабільним пулом Fe2+ та пероксидацією ліпідів, провокує загибель клітин через руйнацію лізосом та мітохондрій [38]. Усі ці процеси відіграють вирішальну роль у пошкодженні міокарда внаслідок хронічної, гострої ішемії, запальних чи інших уражень міокарда, визначають втрату кардіоміоцитів з формуванням хронічної серцевої недостатності при різних серцевих захворюваннях.
Разом з тим в організмі людини існують антиоксидантні системи, які протидіють агентам окисного стресу, у фізіологічних умовах утворення активних форм кисню врівноважується їх елімінацією, що підтримує рівень окиснювачів, достатній для виконання фізіологічних функцій як сигнальних молекул. Баланс згаданих систем обумовлює виживання чи загибель клітин. Антиоксиданти можуть працювати як безпосередні елімінатори молекул АФК, а також через модуляцію активованих окиснювачами процесів загибелі клітин. До антиоксидантів належать низка ферментних (супероксиддисмутази, каталази, глутатіонпероксидази, пероксиредоксини та тіоредоксини) та неферментних молекул (вітаміни Е та С, сечова кислота тощо), а також регуляторні системи, які відповідають за адаптацію тканин в умовах окисного стресу [27, 29]. Зокрема, такими системами є сигнальні шляхи, пов’язані з фактором транскрипції NF-κB, що регулює роботу генів, відповідальних за виживання клітин, та з ядерним фактором Nrf2, що відповідає за активацію антиоксидантних систем. Фактор транскрипції NF-κB підвищує експресію апоптотичних білків. Білок Nrf2 знаходиться в цитозолі під контролюючим впливом молекули KEAP1, яка потенціює деградацію білка. Накопичення АФК викликає руйнування зв’язку у макромолекулі Nrf2-KEAP1, активований Nrf2 переміщується в ядро, де функціонує як фактор транскрипції, зв’язується з доменом ARE та призводить до експресії антиоксидантних генів з активацією глутатіонпероксидази, гемоксигенази-1 та інших важливих компонентів антиоксидантної системи [32]. Отже, елімінація АФК може запобігти як розвитку, так і прогресуванню кардіоваскулярної патології, а застосування антиоксидантів справедливо розглядається як дієва терапевтична стратегія.
Особливості ішемічно-реперфузійного пошкодження міокарда
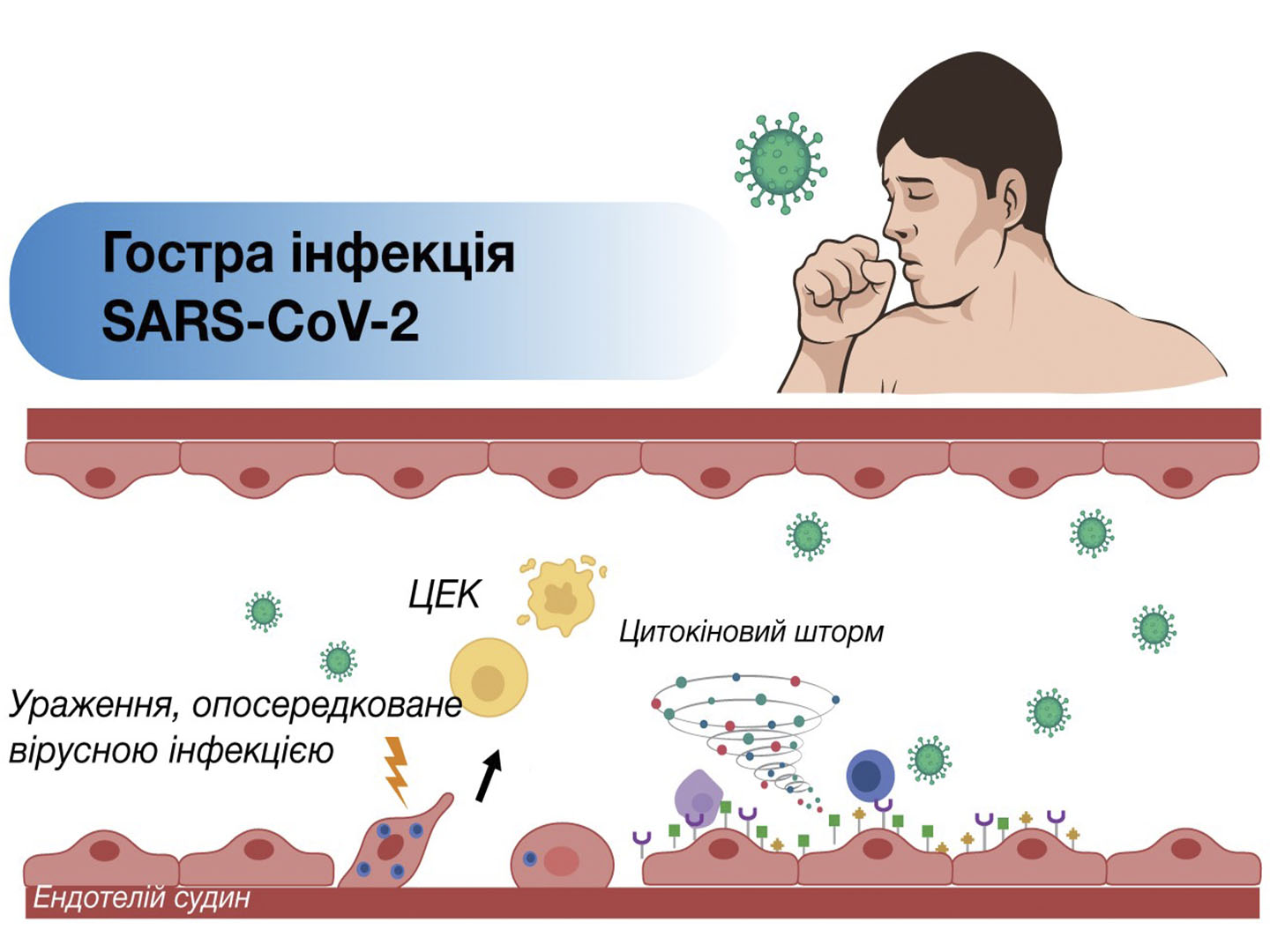
Особливу роль окисний стрес відіграє при гострому коронарному синдромі (ГКС). З прогресивним запровадженням реперфузійної терапії, перкутанних коронарних втручань та регулярного вдосконалення заходів вторинної профілактики у пацієнтів з гострим коронарним синдромом пов’язують загальну тенденцію до зниження смертності від ІХС, яка намітилася у Європі за останні тридцять років. Зрозуміло, що застосування прямих дієвих способів відновлення коронарного кровотоку в ішемізованій ділянці міокарда повинно призвести до покращення як безпосередніх, так і віддалених наслідків ГКС. Широким колом клінічних досліджень та багаторічною практикою підтверджено, що успішне раннє коронарне втручання суттєво обмежує розмір інфаркту, запобігає шлуночковій дисфункції та розвитку фатальних ускладнень. Проте доводиться визнати, що на додаток до очевидних переваг такого втручання можна отримати реперфузійне пошкодження міокарда (РПМ).
Уперше феномен описали в експериментальній моделі інфаркту міокарда в 1960 році Jennings et al. [33]. Загалом наслідки ГКС обумовлені як ішемією міокарда через гостру коронарну оклюзію, так і процесами, які може запустити відновлення кровотоку. Реперфузію міокарда, спонтанну чи як результат активного лікування ГКС, влучно назвали «двосічним мечем», оскільки майже одразу після відновлення кровотоку запускається цілий каскад небажаних явищ, у порочному колі яких надмірне утворення активних форм кисню є ключовим, проте не єдиним фактором, що може призводити до додаткового пошкодження і загибелі кардіоміоцитів [27]. Найчастіше РПМ реалізується в електрофізіологічну нестабільність з розвитком реперфузійних аритмій, систолічну дисфункцію – «оглушення» міокарда з механічною контрактильною дисфункцією, ендотеліальну та мікросудинну дисфункцію – феномен no-reflow, необоротне пошкодження міокарда через прискорення загибелі кардіоміоцитів. Реперфузійне пошкодження завершується смертю тих кардіоміоцитів, які були життєздатними безпосередньо перед реперфузією міокарда, і таким чином, за даними низки авторів, може бути відповідальним за 50 % кінцевого розміру зони некрозу [24]. Саме феномен РПМ може пояснити, чому, незважаючи на оптимальну реперфузію міокарда, відсоток смертей після гострого інфаркту міокарда наближається до 10 %, а частота розвитку серцевої недостатності становить майже 25 % [32]. Для розуміння шляхів подолання реперфузійного пошкодження міокарда необхідно враховувати його механізми, які між собою тісно взаємопов’язані. На сьогодні основними вважаються такі процеси: а) утворення активних форм кисню з накопиченням АФК у відповідь на реоксигенацію міокарда – «кисневий парадокс»; б) зменшення оксиду азоту (NO) з ендотеліальною дисфункцією та порушенням регіонарного кровотоку; в) внутрішньоклітинне та мітохондріальне перевантаження Ca2+ – «кальцієвий парадокс» з набряком та некрозом клітин; г) відкриття перехідної пори проникності мітохондрій (mPTP); д) активація запальної відповіді [3, 9, 17, 24].
Вирішальну роль у цьому сценарії відіграє окисний стрес. Разом з пошкодженням зовнішніх та внутрішніх мембран клітини перекисним окисненням ліпідів Ca2+-перевантаження та відкриття mPTP веде до масивного набряку, лізису органел, до фрагментації та некрозу клітин, що запускає гостру запальну відповідь, наслідки якої також роблять свій внесок у загибель кардіоміоцитів [12]. Через кілька годин після початку реперфузії у зону інфаркту активно мігрують нейтрофіли у відповідь на вивільнення хемоатрактантів (АФК, цитокінів і активованого комплементу). Масивне накопичення нейтрофілів сприяє вивільненню NF-κB, інших сигнальних молекул та факторів транскрипції, посиленій експресії молекул клітинної адгезії, що веде до подальшої лейкоцитарної інфільтрації, закупорки судин та феномену no-reflow, посилюючи пошкодження тканин [27, 33]. Крім того, пероксинітрит дезактивує ендотеліальну NO-синтазу, що сприяє розвитку ендотеліальної дисфункції, що поглиблює порушення регіонарного кровотоку.
Як зазначалося вище, надлишок АФК активує також процеси відтермінованої смерті кардіоміоцитів – апоптозу та фероптозу, у тому числі через активацію сигнальних шляхів, які покликані підвищувати адаптацію тканин до умов окисного стресу та є частиною антиоксидантної системи нашого організму. Низка досліджень показали, що апоптотичний компонент загибелі клітини або запускається, або прискорюється саме під час реперфузії, а не у період ішемії [32]. Zhao et al. пояснюють це тим, що апоптоз є енергозалежним процесом, а рівень АТФ у клітинах значно знижується при ішемії і поповнюється при відновленні кровотоку внаслідок реперфузії [40].
Ці ж автори в експериментальному дослідженні продемонстрували, що некротична загибель клітин досягає піку через 24 години після реперфузії, а апоптотична загибель клітин має максимум до 72 годин після реперфузії. Отже, фармакологічна блокада сигнальних каскадів програмованої загибелі клітин, поряд з елімінацією АФК, під час реперфузії може бути перспективною терапевтичною мішенню у профілактиці наслідків реперфузійного пошкодження міокарда. Цей висновок знайшов своє підтвердження в експерименті, під час якого інгібування індукованого реперфузією апоптозу призвело до зменшення розміру інфаркту та покращення скоротливої функції міокарда [17].
Таким чином, з огляду на відомі механізми взаємодії активних форм кисню та антиоксидантних систем і роль балансу між ними у патогенезі кардіоваскулярної патології, терапевтичних мішеней у боротьбі з наслідками окисного стресу може бути декілька, зокрема елімінація окиснювачів, активація сигнальних систем продукції антиоксидантів, модуляція процесів програмованої загибелі клітин. Ідеально, якщо антиоксидантний засіб може втручатися на усіх вказаних рівнях, працювати як всередині клітини, так і у міжклітинному просторі.
Перспективи застосування едаравону для профілактики реперфузійного пошкодження міокарда
Історія використання антиоксидантів для лікування серцево-судинної патології досить довга, разом з тим результати клінічних досліджень щодо ефективності їх застосування як кардіопротекторів при ішемії, зокрема для профілактики реперфузійного пошкодження міокарда, досить скромні [3, 4]. Едаравон є найбільш перспективним антиоксидантом завдяки своїм особливостям: має низьку молекулярну масу, є водорозчинним та ліпофільним, може залишатися в організмі у дієвій концентрації до 12 годин [2, 28]. Крім того, завдяки багатогранним ефектам препарат має широкий вплив на різні ланки складного патогенезу ішемічного каскаду.
Розглянемо основні ефекти едаравону, про які відомо на сьогодні. В основі антиоксидантної дії препарату лежить його здатність віддавати електрон різним формам АФК, у першу чергу пероксильному радикалу, що розриває ланцюг окиснення ліпідів у мембранах клітин та ліпопротеїдів у системному кровотоці, зменшує концентрацію гідроксильних та пероксильних радикалів в умовах експериментально змодельованої ішемії [26]. Відомо, що ліпідні радикали дезактивують ендотеліальну NO-синтазу (eNOS), а дефіцит оксиду азоту порушує функцію ендотелію, сприяє атеросклеротичному ураженню. Натомість едаравон, знижуючи рівень окиснення ЛПНЩ, стабілізує мРНК NO-синтази, відновлює її експресію на ендотеліальних клітинах, що, у свою чергу, підвищує синтез оксиду азоту, відновлює функцію ендотелію та може захистити тканини в умовах ішемії [28].
Загалом щодо до пулу NO-синтаз препарат має регулюючий ефект, особливо щодо індуцибельної NO-синтази (iNOS), яка продукується макрофагами та нейтрофілами в рамках запальної відповіді та сприяє утворенню пероксинітриту – однієї з найагресивніших форм АФК. Було показано, що застосування едаравону в моделі ішемічного інсульту веде до пригнічення активності iNOS та, як наслідок, зменшення утворення пероксинітриту, що захищало мозкову тканину, зменшувало її набряк та інші прояви запалення, покращувало мозковий кровотік [18]. Ще один корисний ефект препарату – стабілізуючий вплив на мітохондрії та на перехідну пору проникності мітохондрій (mPTP), що може захистити клітину від перевантаження Са2+, зокрема, в умовах «кальцієвого парадоксу» при реперфузійному пошкодженні міокарда [35]. Саме mPTP є досить перпективною терапевтичною мішенню, оскільки ця структура не тільки регулює метаболізм клітин у різних умовах функціонування, але і визначає реалізацію мітохондріального сигнального шляху апоптозу [24, 34]. Тобто застосування едаравону може захищати клітини різних тканин і від надмірного апоптозу. Реалізується цей захист шляхом пригнічення експресії різноманітних протеїнів, залучених до активації апоптозу, зокрема Fac-білка з домену смерті та HMGB-1 білка, з вивільненням якого пов’язують масивне пошкодження ішемізованих тканин [16]. В умовах змодельованого інфаркту міокарда едаравон пригнічує апоптоз і через деактивацію янус-кіназного (JAK-STAT3) сигнального шляху [5, 12, 39]. Важлива складова ефекту препарату – активація природних антиоксидантних систем у тканинах. Так, в експерименті з ішемією міокарда в пацюків попереднє введення едаравону призводило до суттєвого підвищення активності таких ферментів-антиоксидантів, як супероксиддисмутаза, глутатіонпероксидаза, каталаза. Також виявилося, що у групі едаравону значно знизився рівень каспази-3, яка є частиною шляху активації апоптозу, а у кардіоміоцитах збереглася нормальна архітектура мітохондрій та інших структур, при цьому низький рівень КФК-МВ свідчив про загалом мінімальну руйнацію міокарда [11]. Едаравон продемонстрував свої позитивні ефекти щодо іншого виду програмованої загибелі клітин, що провокується окисним стресом, – фероптозу. Було показано, що застосування препарату нормалізує такі характерні для фероптотичних клітин метаболічні зрушення, як накопичення Fe2+ та високий рівень ліпідних та гідроксильних радикалів [14, 38]. Відбувається це шляхом прямої донації електронів та нейтралізації вільних радикалів, а також шляхом активації сигнального білка Nrf2, який запускає процеси ядерної транскрипції з синтезом ферментів глутатіонпероксидази (Gpx4) та гемоксигенази-1, що руйнують АФК та пригнічують фероптоз. Такий шлях захисного ефекту едаравону підтверджений експериментально на різних тваринних моделях у багатьох дослідженнях in vitro [36].
Окисний стрес тісно пов’язаний і з процесами запалення: з одного боку, запалення супроводжується утворенням активних форм кисню, з іншого боку, ліпідні та пероксидрадикали активують ядерний фактор NF-κB та інші посередники запалення, зокрема iNOS, про яку йшлося вище. На кроликовій моделі атеросклерозу показано, що едаравон здатний пригнічувати активацію фактора NF-κB та iNOS і зменшувати таким чином надмірну міграцію макрофагів у зону бляшки, проліферацію гладкої мускулатури, що разом з прямою його антиоксидантною активністю та позитивним впливом на дисфункцію ендотелію через еNOS веде до зменшення атеросклеротичного ураження аорти [25]. Інші дослідники продемонстрували, що, очевидно, через нормалізацію функції ендотеліальних клітин з активацією тканинного активатора плазміногену едаравон сприяє швидкій реканалізації оклюзованої тромбом судини, а отже, препарат має підтверджений позитивний вплив на перебіг атеросклеротичного процесу на усіх його етапах [21].
Таким чином, едаравон можна вважати універсальним та багатогранним антиоксидантом, який має ще й цитопротекторний ефект, захищаючи клітини від кількох видів загибелі: безпосередньої – некрозу, відтермінованих – апоптозу та фероптозу, демонструє також протизапальний ефект та позитивно впливає на судинну стінку. Усі ці ефекти вкрай корисні при серцево-судинних захворюваннях, зокрема при ГКС, у патогенезі якого окисний стрес, запалення, різні типи загибелі клітин відіграють основну роль та визначають наслідки гострого ішемічного пошкодження міокарда.
Експериментальне застосування едаравону на тваринних моделях гострої ішемії міокарда підтвердило його захисні властивості [8, 25]. Введення препарату на молекулярному рівні супроводжувалося пригніченням пероксидації ліпідів, нормалізацією вмісту АТФ, підвищенням рівня NO, зменшенням Са2+-перевантаження, пригніченням утворення TNF-α, сигнальних білків апоптозу, на клітинному рівні – зменшенням кількості пошкоджених кардіоміоцитів з фрагментованими органелами та фатальним набряком, апоптотичних клітин [8, 10, 20]. На макрорівні це супроводжувалося зменшенням розміру зони некрозу при ішемії/реперфузії, нормалізацією мікросудинної ендотеліальної дисфункції, профілактувало розвиток фатальних шлуночкових тахіаритмій та подальший розвиток чи прогресування серцевої недостатності. Такі захисні ефекти едаравон демонстрував при застосуванні до або одразу після (не пізніше 5 хвилин) початку реперфузії [10].
Пілотне рандомізоване клінічне дослідження едаравону у пацієнтів з інфарктом міокарда провели ще у 2004 році японські науковці. Було залучено 80 пацієнтів, препарат вводили у дозі 30 мг за 10 хвилин до початку перкутанного втручання [30]. Тоді вперше підтвердили ефективність превентивного застосування едаравону саме для профілактики реперфузійних аритмій під час інтервенційного лікування ГКС. Також було показано, що застосування препарату до реперфузійних процедур супроводжується вірогідним зменшенням зони некрозу зі зниженням рівня маркерів пошкодження міокарда, зменшенням «оглушення» міокарда зі швидким відновленням його скоротливості [30]. У наступному дослідженні з більшою вибіркою (101 пацієнт) Tsujita et al. [31] продемонстрували, що пацієнти з групи едаравону порівняно з групою плацебо мали значно вищий кумулятивний показник без фатальних подій у віддалений після ГКС період. За даними авторів, застосування препарату у гострий період протягом 14 діб захищає від подальших кардіоваскулярних подій (повторного нефатального інфаркту, ішемічного інсульту, рефрактерної стенокардії, період спостереження 415 ± 32 дні). Едаравон продемонстрував свою ефективність серед пацієнтів з ГКС у зменшенні зони інфаркту та профілактиці реперфузійних аритмій у разі його застосування до проведення реперфузії у перші 6 годин після виникнення симптомів.
Надалі у декількох клінічних дослідженнях також було підтверджено, що включення препарату до схеми лікування хворих з інфарктом міокарда у поєднанні з тромболітичною терапією зменшує зону некрозу, покращує фракцію викиду лівого шлуночка, зменшує ризик фатальних тахіаритмій, зменшує ймовірність повторної госпіталізації у зв’язку з декомпенсацією СН [6, 13, 15]. Nakamura et al. [23] показали, що застосування едаравону веде до зменшення зони некрозу та запобігає патологічному ремоделюванню міокарда з порушенням його функції та формуванням СН, у тому числі шляхом нівелювання запальної складової РПМ. Oyama et al. [26] повідомили, що введення препарату пацієнтам зі значимим стенозом коронарних артерій супроводжувалося вірогідним покращенням коронарного кровотоку (за даними коронарографії) та суттєвим зниженням рівня малональдегіду (маркера перекисного окиснення ліпідів).
За останнє десятиліття дані щодо клінічного застосування едаравону при інфаркті міокарда продовжують накопичуватися. Ґрунтовне узагальнення відомостей щодо ефективності препарату при гострій ішемії міокарда подали китайські науковці у метааналізі 2015 року [41]. До резюме включили результати 9 однорідних рандомізованих плацебо-контрольованих досліджень, хоча, як повідомлять науковці, у різних наукових базах даних було знайдено 135 публікацій за темою. У дослідження було залучено загалом 380 пацієнтів; досліджуваний препарат застосовували у дозі 60 мг на добу. Основною метою метааналізу було знайти підтвердження механізмів кардіоцитопротекторного та антиоксидантного ефекту препарату у пацієнтів з ГКС, які перенесли перкутанне втручання. Встановили, що у групах едаравону спостерігалося вірогідне зменшення ензиматичного розміру інфаркту за зниженням рівня маркерів некрозу міокарда КФК-МВ та тропоніну І у 3 дослідженнях через 6 годин після реперфузії, а у 4 дослідженнях – через 24 години, на відміну від групи плацебо. Причому вплив на рівень тропоніну І був дозозалежний, найнижчі концентрації маркера некрозу визначалися у разі введення 100 мг препарату. При аналізі маркерів окисного стресу виявилося, що на тлі застосування едаравону рівень супероксиддисмутази зростав, а концентрація малон-альдегіду (маркера перекисного окиснення ліпідів) зменшувалася на 6-й годині у 4 дослідженнях та на 24-й годині у 7 та 8 дослідженнях. Таким чином, метааналіз переконливо підтвердив, що едаравон працює як антиоксидант, а його кардіопротекторні можливості чітко пов’язані з позитивними клінічними ефектами у запобіганні реперфузійному пошкодженню міокарда [41].
При ГКС рекомендовано застосування едаравону за схемою: першу дозу едаравону (30 мг) вводять внутрішньовенно струминно протягом 10 хвилин перед реперфузією, далі вводять по 30 мг у розведенні на 100 мл 0,9% розчину натрію хлориду внутрішньовенно краплинно двічі на добу протягом 14 діб.
Таким чином, едаравон є потужним антиоксидантом з додатковими антиапоптотичним та антифероптотичним, антинекротичним, протизапальним ефектами, стабілізує кардіоміоцити та ендотеліальні клітини, а його призначення при гострому коронарному синдромі є перспективною стратегією профілактики реперфузійного пошкодження зі зменшенням зони некрозу, профілактикою фатальних порушень ритму, патологічного ремоделювання міокарда та судин. Завдяки своїм багатогранним ефектам едаравон може бути корисним і при інших кардіоваскулярних захворюваннях.
Конфлікт інтересів. Не заявлений.
Автори: Жебель В.М., Старжинська О.Л. Вінницький національний медичний університет ім. М.І. Пирогова, м. Вінниця, Україна.
Для кореспонденції: Жебель Вадим Миколайович, д.м.н., професор, завідувач кафедри внутрішньої медицини медичного факультету № 2, Вінницький національний медичний університет ім. М.І. Пирогова, вул. Пирогова, 56, м. Вінниця, 21018, Україна; e-mail: redact@i.ua.
Література:
- Палаткина Л.О., Корнеева О.Н., Драпкина О.М. Окислительный стресс – роль в патогенезе хронической сердечной недостаточности, возможности коррекции. Кардиоваскулярная терапия и профилактика. 2012. 11(6). 91-94. https://doi. org/10.15829/1728-8800-2012-6-91-94
- Ali Z.K., Baker D.E. Formulary drug review: edaravone. Hospital pharmacy. 2017. 52(11). 32-736. https://doi.org/10.1177/ 0018578717734877
- Bainey K.R., Armstrong P.W. Clinical perspectives on reperfusion injury in acute myocardial infarction. American heart journal. 2014. 167(5). 637-645. doi.org/10.1016/j.ahj.2014.01.015
- Casas A.I., Nogales C., Mucke H.A., Petraina A., Cuadrado A., Rojo A.I. et al. On the clinical pharmacology of reactive oxygen species. Pharmacological reviews. 2020. 72(4). 801-828. DOI: https://doi.org/10.1124/pr.120.019422
- Chen H., Chen Y., Wang X., Yang J., Huang C. Edaravone attenuates myocyte apoptosis through the JAK2/STAT3 pathway in acute myocardial infarction. Free Radic. Res. 2020. 54(5). 351-359. doi: 10.1080/10715762.2020.1772469.
- Cui Y.G., Lu X.N. Edaravone on reperfusion injury of acute myocardial infarction. Journal of Clinical Medicine in Practice. 2011. 3. 71-73.
- ESC Scientific Document Group, 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). European Heart Journal. 2021. 42(14). 1289-1367. https:// doi.org/10.1093/eurheartj/ehaa575
- Fukuda A., Okubo S., Tanabe Y., Hoshiba Y., Shiobara H., Harafuji K. et al. Cardioprotective effect of edaravone against ischaemia-reperfusion injury in the rabbit heart before, during and after reperfusion treatment. Journal of international medical research. 2006. 34(5). 475-484. https://doi.org/10.1177/147323000603400504
- González-Montero J., Brito R., Gajardo A.I., Rodrigo R. Myocardial reperfusion injury and oxidative stress: Therapeutic opportunities. World J. Cardiol. 2018. 10(9). 74-86. DOI: 10.4330/wjc.v10.i9.74
- Hassan M.Q., Akhtar M.S., Akhtar M., Ali J., Haque S.E., Najmi A.K. Edaravone protects rats against oxidative stress and apoptosis in experimentally induced myocardial infarction: Biochemical and ultrastructural evidence. Redox Report. 2015. 20(6). 275-281. https://doi.org/10.1179/1351000215Y.0000000011
- Hassan M.Q., Akhtar M.S., Akhtar M., Ali J., Haque S.E., Najmi A.K. Edaravone, a potent free radical scavenger and a calcium channel blocker attenuate isoproterenol induced myocardial infarction by suppressing oxidative stress, apoptotic signaling and ultrastructural damage. Therapeutic advances in cardiovascular disease. 2016. 10(4). 214-223. doi.org/10.1177/1753944716630 653.
- Hassan M.Q., Akhtar M.S., Afzal O., Hussain I., Akhtar M., Haque S.E., Najmi A.K. Edaravone and benidipine protect myocardial damage by regulating mitochondrial stress, apoptosis signalling and cardiac biomarkers against doxorubicin-induced cardiotoxicity. Clin. Exp. Hypertens. 2020. 42(5). 381-392. doi: 10.1080/10641963. 2019.1676770. Epub 2019 Oct 20.
- Higashi Y., Jitsuiki D., Chayama K., Yoshizumi M. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases. Recent Patents on Cardiovascular Drug Discovery (Discontinued). 2006. 1(1). 85-93.
- Homma T., Kobayashi Sh., Sato H., Fujii J. Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Experimental Cell Research. 2019. 384(1). 111592. https://doi. org/10.1016/j.yexcr.2019.111592
- Hou H. Study on the Protective Effect of Edaravone on Myocardial Ischemia Reperfusion Injury. Advanced Emergency Medicine. 2013. 2(1). 10-12.
- Ikegami E., Fukazawa R., Kanbe M., Watanabe M., Abe M., Watanabe M., et al. Edaravone, a potent free radical scavenger, prevents anthracycline-induced myocardial cell death. Circulation Journal. 2007. 71(11). 1815-1820. https://doi.org/10.1253/ circj.71.1815
- Kalogeris Th., Bao Y., Korthuis R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biology. 2014. 2. 702-714. https://doi. org/10.1016/j.redox.2014.05.006
- Kikuchi K., Tancharoen S., Takeshige N., Yoshitomi M., Morioka M., Murai Y., Tanaka E. The efficacy of edaravone (radicut), a free radical scavenger, for cardiovascular disease. International journal of molecular sciences. 2013. 14(7). 13909-13930. doi.org/10.1179/1351000215Y.0000000011
- Kiyuna L.A., e Albuquerque R.P., Chen C.H., Mochly-Rosen D., Ferreira J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: challenges and opportunities. Free Radical Biology and Medicine. 2018. 129. 155-168. doi.org/10.1016/j.freeradbiomed.2018.09.019
- Li Q., Qiu Z., Lu Y., Lu P., Wen J., Wang K. et al. Edaravone protects primary-cultured rat cortical neurons from ketamine-induced apoptosis via reducing oxidative stress and activating PI3K/Akt signal pathway. Molecular and Cellular Neuroscience. 2019. 100. 103399. https://doi.org/ 10.1016/j.mcn.2019.103399
- Mankar V.H. Edaravone based antioxidants for cardioprotection and neuroprotection. Plant. Archives. 2020. 20(2). 3440-3445.
- Murray Ch.J.L., Aravkin A.Y., Zheng P., Abbafati C., Abbas K.M., Abbasi-Kangevari M. et al. Global burden of disease 2019 risk factor Collaborators. Lancet. 2020. 396. 1135-59. https://doi.org/10.1016/S0140-6736(20)30752-2
- Nakamura Y., Yamada Y., Shimomura H., Nagayoshi Y., Tsujita K., Yamashita T. et al. The effect of edaravone on plasma monocyte chemoattractant protein-1 levels in patients with acute myocardial infarction. Journal of cardiology. 2009. 54(3). 416-424. https://doi.org/10.1016/j.jjcc.2009.07.001
- Neri M., Riezzo I., Pascale N., Pomara C., Turillazzi E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediators Inflamm. 2017. 2017. 7018393. doi: 10.1155/2017/7018393
- Onogi H., Minatoguchi S., Chen X.H., Bao N., Kobayashi H., Misao Y. et al. Edaravone reduces myocardial infarct size and improves cardiac function and remodelling in rabbits. Clinical and experimental pharmacology and physiology. 2006. 33(11). 1035-1041. https://doi.org/10.1111/j.1440-1681.2006.04483.x
- Oyama J., Satoh S., Suematsu N., Kadokami T., Maeda T., Sugano M., Makino N. Scavenging free radicals improves endothelial dysfunction in human coronary arteries in vivo. Heart Vessels. 2010. 25(5). 379-85. doi: 10.1007/s00380-009-1221-7
- Pagliaro P., Moro F., Tullio F., Perrelli M.-G., Penna C. Cardioprotective Pathways During Reperfusion: Focus on Redox Signaling and Other Modalities of Cell Signaling. Antioxidants & Redox Signaling. 2011. 3. 833-850. http://doi.org/10.1089/ars.2010.3245
- Subcommittee U.P., Advantage M., Plan M.S.N., Select C. Required Clinical Documentation for Review. Policy. 2020. 10. 12.
- Tao Xu, Wei Ding, Xiaoyu Ji, Xiang Ao, Ying Liu, Wanpeng Yu, Jianxun Wang. Oxidative Stress in Cell Death and Cardiovascular Diseases. Oxidative Medicine and Cellular Longevity. 2019. Article ID 903056311. https://doi.org/10.1155/2019/9030563
- Tsujita K., Shimomura H., Kawano H., Hokamaki J., Fukuda M., Yamashita T. et al. Effects of edaravone on reperfusion injury in patients with acute myocardial infarction. The American Journal of Cardiology. 2004. 94(4). 481-484. doi: 10.1016/j.amjcard.2004.05.007
- Tsujita K., Shimomura H., Kaikita K., Kawano H., Hokamaki J., Nagayoshi Y. et al. Long-term efficacy of edaravone in patients with acute myocardial infarction. Circulation journal. 2006. 70(7). 832-837. https://doi.org/10.1253/circj.70.832
- Xiang M., Lu Y., Xin L., Gao J., Shang C., Jiang Z. et al. Role of Oxidative Stress in Reperfusion following Myocardial Ischemia and Its Treatments. Oxid. Med. Cell. Longev. 2021. 2021. 6614009. doi: 10.1155/2021/6614009. PMID: 34055195. PMCID: PMC8149218.
- Yellon D.M., Hausenloy D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007. 357. 1121-1135.
- Zhang G.M., Wang Y., Li X.Y., Xu L., Su S.P., Sun Y.Y. Postconditioning of lactic acid and edaravone reduces apoptosis of myocardiocytes through the p38/JNK pathway. Chinese Journal of Geriatric Heart Brain and Vessel Diseases. 2013. 7. 753-757.
- Zhang G.M., Wang Y., Li X.Y., Xu L., Sun Y.Y., Li Y.Z., Liu L.F. Pharmacological postconditioning with lactic acid and low dose edaravone could attenuate myocardial reperfusion injury through mitochondrial pathway. Zhonghua Xin Xue Guan Bing Za Zhi. 2013. 41(8). 647-53. PMID: 24225235.
- Zhang G.W., Gu T.X., Sun X.J., Wang C., Qi X., Wang X.B., Li-Ling J. Edaravone promotes activation of resident cardiac stem cells by transplanted mesenchymal stem cells in a rat myocardial infarction model. The Journal of thoracic and cardiovascular surgery. 2016. 152(2). 570-582. doi.org/10.1016/j.jtcvs.2016.02.071
- Zhang W.-W., Bai F., Wang J., Zheng R.-H., Yang L.-W., James E.A., Zhao Z.-Q. Edaravone inhibits pressure overload-induced cardiac fibrosis and dysfunction by reducing expression of angiotensin II AT1 receptor. Drug Des. Dev. Ther. 2017. 1. 3019-3033. doi.org/10.2147/DDDT.S144807
- Zhang Y., Xin L., Xiang M., Shang C., Wang Y., Wang Y., Cui X., Lu Y. The molecular mechanisms of ferroptosis and its role in cardiovascular disease. Biomed Pharmacother. 2021. 145. 112423. doi: 10.1016/j.biopha.2021.112423
- Zhao X., Zhang E., Ren X. et al. Edaravone alleviates cell apoptosis and mitochondrial injury in ischemia-reperfusion-induced kidney injury via the JAK/STAT pathway. Biol. Res. 2020. 53. 28. https://doi.org/10.1186/s40659-020-00297-0
- Zhao Z.Q., Velez D.A., Wang N.P., Hewan-Lowe K.O., Nakamura M., Guyton R.A. et al. Progressively developed myocardial apoptotic cell death during late phase of reperfusion. Apoptosis. 2001. 6(4). 279-290.
- Zheng C., Liu S., Geng P., Zhang H., Zhang H., Tang A., Xie X. Efficacy of edaravone on coronary artery bypass patients with myocardial damage after ischemia and reperfusion: a meta analysis. International journal of clinical and experimental medicine. 2015. 8(2). 2205-2211.